Oil market watchers always keep half an eye on weather forecasts. However, over the past few months, their attention has been more concentrated. As an unusually ferocious hurricane season has buffeted the US and its key producing regions, questions have been asked not only about the short-term disruption caused but also about the responsibility of the downstream industry - rather than OPEC and its counterparts - for prices that, while easing, remain at a level more than twice what producers once pronounced as their ideal. The target OPEC price band of $22-28 a barrel seems a distant memory.
Spot Brent was trading at more than $50 a barrel in mid-November. And this despite most market factors being bearish: Americans are hunkering down for the winter in unusually mild temperatures; upstream and downstream facilities in the Gulf of Mexico are gradually recovering from the worst of the hurricanes, Katrina and Rita; and stock levels are reasonable for the time of year. 'US industry stocks have increased [by] 18.5 million barrels since end-September,' says Geoff Pyne, analyst at ABN Amro. 'The weather in both the US and Europe has been mild in recent weeks so one of the market's main concerns, winter distillate stocks, do not hold the threat they appeared to. US distillate stocks are at the low end of their seasonal range but Europe has a growing overhang of supplies available for export.' But as oil prices settle at a level that a couple of years ago would have appeared outrageous, the debate continues as to responsibility. The argument has even reached Capitol Hill, where the oil majors have been called on to explain why windfall profits have not been used to ease local consumers' gasoline bills. And OPEC is becoming increasingly outspoken in blaming high prices on downstream bottlenecks and lack of investment to clear them, as the producer group collectively pumps well above its theoretical 28 million-barrel-a-day (b/d) production ceiling - due for review at a meeting in Kuwait on 12 December (see table). 'The primary responsibility for refining capacity expansion remains with the major consuming countries,' OPEC said in a recent report. 'OPEC has called on these countries to create the appropriate environment and provide the necessary incentives to ensure timely and sufficient investments in this key sector. While the role of the international oil companies [IOCs] will be crucial to this effort, recent increases in revenue have not been translated into substantial additional investments, despite the needs of the downstream sector.' Speaking at a conference in Moscow in mid-November, OPEC's acting secretary-general Adnan Shihab-Eldin drove home the point: 'Concrete measures should be taken on the part of consuming countries to create an enabling environment to encourage rapid, sizeable investments in the refining sector, especially in conversion capacity, which has persistently lagged behind market requirements,' he said. It is a view that has been repeated many times over the past few months, in response to the complaints of consuming countries about the detrimental effect on their economic growth of oil prices consistently pushing or breaking the $60-a-barrel mark. OPEC ministers insist the market is well supplied with crude and that factors beyond its control - chiefly downstream problems, rampant demand growth in developing countries such as China and India and geopolitical uncertainty in key producing states - are buoying the market. Escalation of tension between Washington and Tehran has increased traders' nervousness, as has the White House's hostility towards key supplier Venezuela. Iraq has been unstable for sufficiently long to be factored out as a political worry, but on a more concrete level, production from the key southern fields was cut in October by poor weather, slashing average monthly output by some 300,000 b/d. 'Aside from the insurgency, the water
You might also like...

Contractors win Oman Etihad Rail packages
23 April 2024
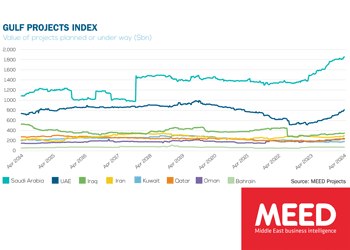
Saudi market returns to growth
23 April 2024
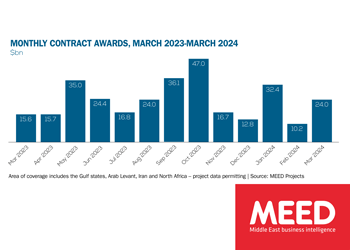
Middle East contract awards: March 2024
23 April 2024

Swiss developer appoints Helvetia residences contractor
23 April 2024




A MEED Subscription...
Subscribe or upgrade your current MEED.com package to support your strategic planning with the MENA region’s best source of business information. Proceed to our online shop below to find out more about the features in each package.